
| | |||||||||||||||||
| |||||||||||||||||
Concepts
- The Sequence alignment
- The Template Structure
- Automatic model building
- Modified bases
- Loops
- Secondary structure
- Base pairing
- Base stacking
- Clash recognition
- Refinement
The Sequence alignment
For template-based modeling, a FASTA file containing two sequences needs to be prepared. The first is the sequence of the model to be built. The second sequence is the template sequence. It needs to be identical with the sequence from the template structure. It can be retrieved by the get_sequence() command. Both need to be aligned in such a way that the structures correspond to each other.
For gaps, dash symbols (-) are used. . may be used to designate unknown bases. For many modified bases, one-letter abbreviations are available (see https://www.genesilico.pl/modomics/modifications. Modifications without a one-letter abbreviation can be represented by "x" or the nomenclature in modomics, e.g. "020U". For all other residues the "." character may be used).
> target sequence GDCGGD--DAGAAUACCUGCCUQUCACGCAGGG-----CGGGTPCGAGU > template sequence GDCGGDCUDAGAAUA----CCUQUCACGCA--GG7UCGCGGGTPCGAGU
The Template Structure
Any RNA structure from the PDB may be used for modeling. The file should be prepared to contain a single model (if not, the first will be taken by default), and alternative conformations should be removed. You should know the chain identifier of the RNA chain you want to use template (A is used by default). Protein chains and non-RNA hetero groups are ignored. ModeRNA uses the remediated PDB nomenclature (e.g. single quotes (‘) instead of asterisks(*) for atom names in the ribose.
Preparation of PDB files
For each RNA template used, it is checked, whether the sequence from the structure is the same as from the corresponding alignment. To check what the exact sequence of your template is, the get_sequence() command may be used.
The most common reason that causes problems while modeling RNA with ModeRNA is that a template is not compatible with the alignment. Possible reasons include:
1. Backbone discontinuity
Symptom:
There are underscore symbols in the sequence.
e.g. structure.get_sequence() ---> 'A_AAAA'
Explanation:
It means that a structures sequence is not continous and can be caused by
some missing backbone atoms, nonproper bond lengths etc. It may also mean
that residues are simply not connected, e.g. when there are two blunt ends of a helix.
You should include the _ in your alignment explicitly e.g.:
A_AAAA A-AGAAModeRNA can try to fix broken backbone with ModeRNA, which helps in many cases. See the
fix_backbone() function for more details.
2. Single ligands, ions or water molecules
Symptom:
There are single underscore-dot symbols at the end of the sequence.
e.g. structure.get_sequence() ---> 'AAAA_.'
Explanation:
It means there is one unidentified residue (ion) at the and of the structure,
that is not connected with the rest of the structure.
This should be included in the alignment (see below) or the ions should be removed from
the template structure using the clean_structure() function.
AAAA_. AAAA--(see 3. if there are more such symbols).
3. Multiple water or ion molecules
Symptom:
There are multiple underscore-dot symbols at the end of the sequence.
e.g. structure.get_sequence() ---> 'AAAA_.'
e.g. structure.get_sequence() ---> 'AAAA_._._._._._._._._._.' (10 water molecules).
Solution:
The water chain should be removed with the clean_structure() function or included in the alignment:
AAAA_._._._._._._._._._. AAAA--------------------
4. Protein chain
Symptom:
The entire chain has a sequence like ._._._. etc.
Solution:
Use a different chain. RNA can't be modeled from proteins yet.
However, the clean_structure() function will remove them.
5. Missing or broken bases.
Symptom:
There are single dots within the sequence.
e.g. structure.get_sequence() ---> 'AA.AAAAA'
Explanation:
It means that one residue is not complete: a whole base or its part is missing
and consequently the residue is marked in the alignment as an unknown residue.
It is not possible to model another base in this place in the automatic mode.
Regardless of used alignment the residue will stay unidentified in the model
(only backbone atoms will be copied to the model).
To be on the safe side, the according residue should be removed manually.
All described above features need to be included in the alignment or the template structure need to be modified.
One may check whether a PDB file (or alignment) needs to be cleaned up.
PDB file modification can be done with the clean_structure() function.
Automatic model building
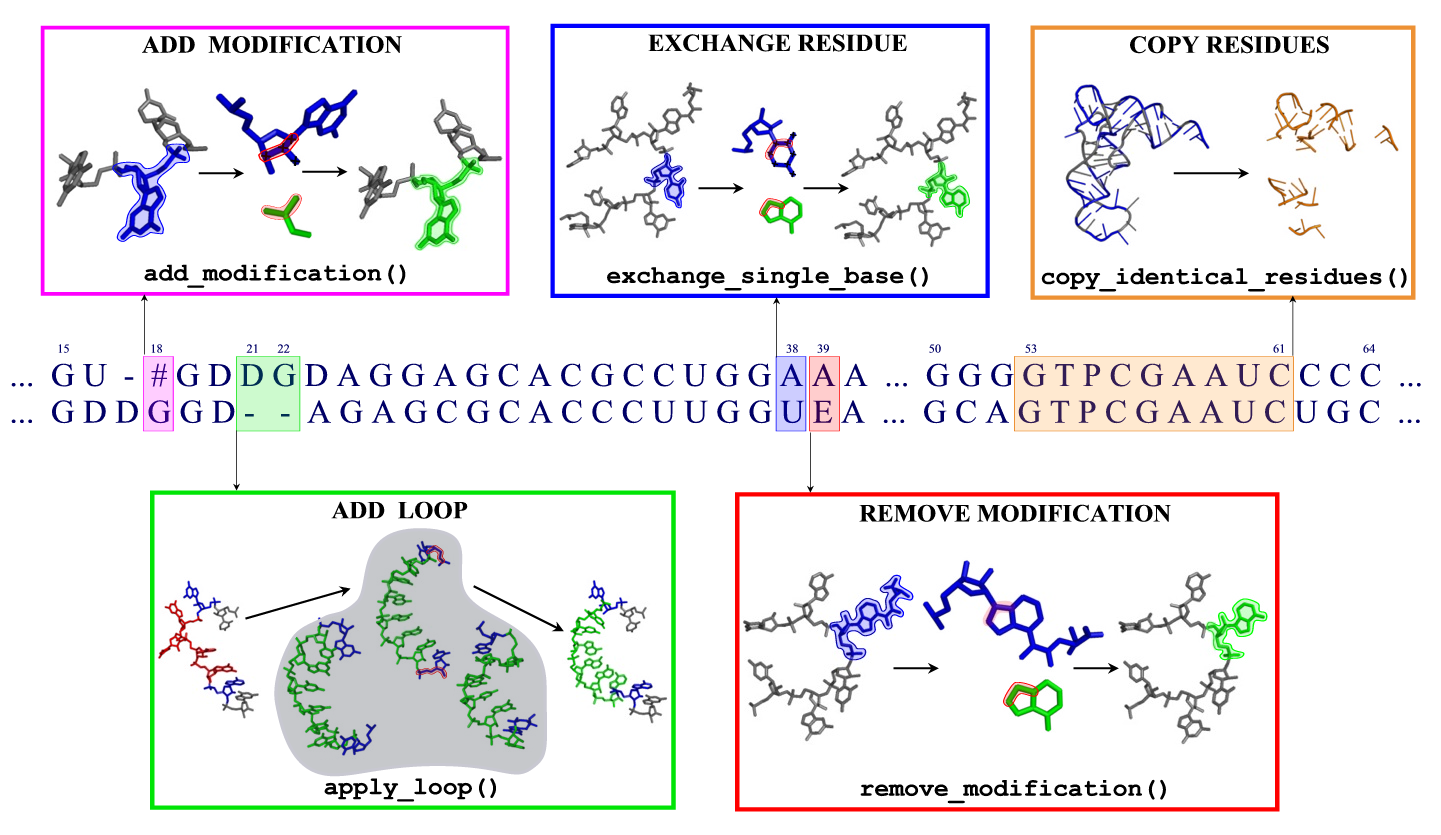
When building a model
- Both letters are identical: All atoms are copied to the model.
- The target contains a different standard base: The base is replaced, retaining the conformation of the glycosidic bond.
- The template or target contains an unknown residue: The backbone and ribose atoms are copied.
- The template is modified but the target is not: Excess atoms belonging to the modification are removed; in bases like pseudouridine, the entire base is replaced. Eventually, the base is replaced afterwards.
- The target is modified, differing from the template: Small molecular fragments are added to construct the modification. Eventually, the base is replaced beforehand (if the modification originates from a different base), and existing modifications are removed.
- There is a gap in either of the sequences: For the entire gap, including two residues on each side, the best fitting fragment is inserted from a library. If two gaps are closer to each other than two positions in the alignment, this cannot be done automatically. See below for details on inserting fragments.
After automatic model building, it is checked, whether the sequences in an alignment and template structure are equal. This includes checking for backbone discontinuities caused by sub-optimal loop insertion. In any case, a detailed report will be written to the moderna.log file.
Modified bases
Moderna is capable of recognizing and editing 115 different base modifications, which have been taken from the Modomics database. For each modification, a name, an abbreviation, a one-letter code, and a molecular structure are given.
A complete list of modification names and abbreviations can be found in Modomics or in the file data/modification_names_table. To mark modifications in the alignment, use the one-letter codes or the new nomenclature column in the table. Example:
> Target containing several modifications as one-letter abbreviations ACUGUGAYUA[U UACCU#PG > Template containing the modification 015U GCGGA----UUU015UALCUCAG
For adding modifications to standard bases, a set of 67 small fragments has been created. Each such fragment contains atoms belonging to a modification, and a triple of connecting atoms which are used to fit the fragment to an existing standard base. These connecting atoms are given in the data/modification_rules file. For removing modifications, a list of atom names which are to be removed is given in the same file.
Recognition of modified nucleotides
The recognition of modified bases is done in two steps: First, if the name of a residue is one out of A,G,C, or U, it is checked whether all and only the atoms for this nucleotide are present. Otherwise, it is assumed that this base is modified. In the second step, modified bases are detected by matching subgraphs in the molecular topology of that residue. For all modifications, such subgraphs have been defined in a SMILE-like notation in such a way, that all of them can be unambiguously distinguished.
This second step is independent of the nomenclature of atoms in the PDB. However, the assignment of the topology of a residue (single and double bonds) is based on interatomic distances. Thus, for a badly resolved or locally distorted template structure, the recognition procedure may fail to recognize a particular residue.
Loops
The loop search procedure in ModeRNA tries to find the best fitting
loop from a library of 88000 RNA fragments. The search is done in two
steps: A fast one based on geometrical parameters, and a slower one
based on the exact fit of a smaller set of loop candidatas. In the
first step, the similarity of seven parameters derived from the
residues at both ends of the fragment is compared to the same
parameters of the residues in the model, between which a linker is to
be found. These parameters involve three distances, two angles, and
three dihedrals (two of which enforce a proper orientation of the
bases). Additionally, the sequence similarity of the linker and the
desired fragment is taken into account. A description of this approach
can be found in
(Michalsky E, Goede A, Preissner R. Loops In Proteins (LIP)
–a comprehensive loop database for homology modelling. Protein Eng. 2003 Dec;16(12):979-85.).
In the second step, the 20 candidate loops that are most similar
according to these criteria are inserted into the model, superimposing
the P,O5',C5' atoms of the terminal residues at the 5' end, and
C4',C3',O3' at the 3' end. A combined score is calculated, using the
RMSD of the superposition, the number of inter-residue clashes, the
number of hydrogen bonds formed by the loop, and the score used in the
first step. The best ranking candidate is then inserted, replacing the
two terminal residues from the model by the loop candidates'.
When building models automatically, ModeRNA eliminates one extra
residue on each side of a gap to be inserted, to give room for a
reasonable loop closure.
Secondary structure
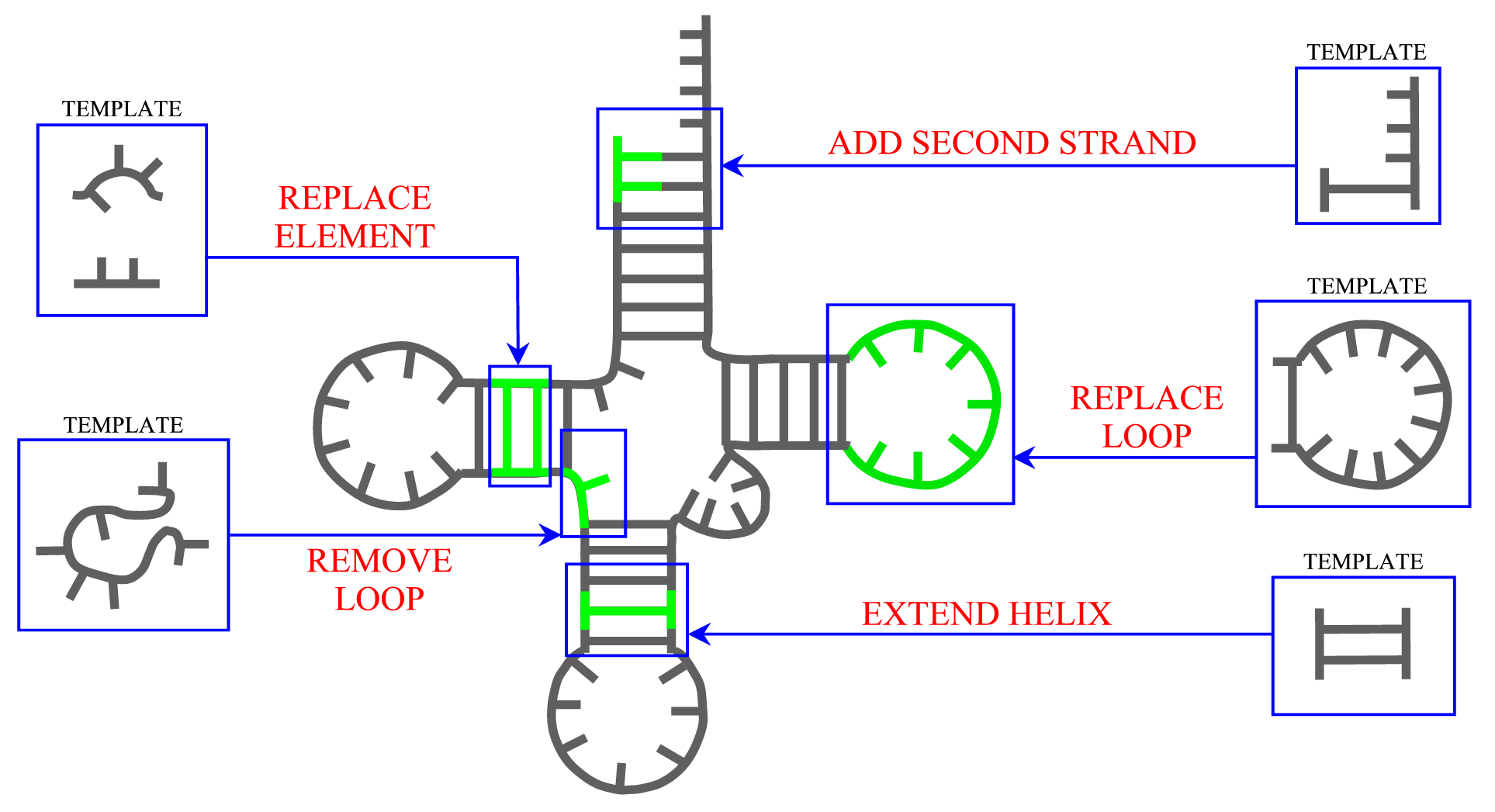
ModeRNA provides a set of functions that can be applied to manipulate secondary structure in the model. The length of helices can be changed and a second strand can be added to a single stranded base. Entire secondary structure fragments provided by a user can be inserted to the structure and, while searching for a missing fragment, ModeRNA can take secondary structure restraints into account. Moreover, the secondary structure can be calculated from the tertiary structure of a model or template. The detailed description of all functions for secondary structure modeling can be found on the Secondary structure page.
Base pairs
The base pairs used by Moderna are calculated according to the Leontis/Westhoff definition [2]. Currently, only the canonical Watson-Crick and Wobble base pairs are used. ModeRNA internally defines three edges in each base (the Watson, Hoogsteen, and Sugar edeg) similar to the RNAView program and tries to recognize the 12 Leontis/Westhof types of noncanonical base pairs. The latter part is however subject to debugging and therefore not publicly advertised (if you are still interested, look for ModernaStructure.get_base_pairs()).
Base stacking
Stacking of bases is calculated according to the procedure described by (Gendron P, Lemieux S, Major F. JMB 2001;308(5):919-36). It essentially defines a range for the angle between the normals of two aromatic rings, their distance, and the horizontal displacement of the rings.
This definition of base stacking is not restricted to adjacent bases, but provides stacking for any two pairs. This procedure has been implemented as part of Moderna. Moderna calculates the base stacking for the template and model structure, to detect differences between the two - in order to detect interactions that were eventually lost during the modeling.
Clash recognition
Interatomics clashes are detected between any pair of atoms that do not belong to the same residue. A clash is defined as an overlap of the atomic radii. The atomic radius again depends on the element.
In Moderna, a set of smaller radii, and considerably bigger VdW radii have been implemented. Which of them is used, is specified in the find_clashes function. The atom radii used can be found in thefollowing table:
The bond between O3’ atom of the preceding and the P atom of the following residue is always exempt from clash recognition. Please note that the hydrogen atoms are not considered, unless they are explicitly contained in the structure.
There exist several definitions of unified atoms, where the hydrogens are represented by a higher atom radius of the attached heavy atom (e.g. the ProtOr set). These radii set conflict with the tightly packed structure of RNA, and lead to clashes even in correct structures. We therefore decided not to use them.
Refinement
Once ModeRNA has produced a model, it may be necessary to refine the local geometry. Mostly, this is the case when for a loop no optimal candidate could be found. In that case, the backbone conformation will contain chemically unreasonable bond lengths, angles and dihedrals. The same may be necessary to optimize the conformation of base pairs. Both issues are addressed by refinement programs.
MMTK is a program library that performs energy minimization using Conjugate Gradient mimimization. It uses the AMBER force field. ModeRNA provides a script to perform minimization of models using MMTK. Alternatively, it is also possible to use a different Molecular Dynamics program like NAMD, or forcefield like Charmm (but for these, no such script exists). Some commercial packages like HyperChem are also capable of performing energy minimization of RNA.
The script refine_model_mmtk.py allows to improve sections of the model using MMTK.
It prepares the structure for input (basically exchanging some residue and atom names),
and starts an energy minimization run. If a range of residues is specified, the ends of
it will be fixed by appying strong distance, angle and torsion restraints.
This way, it is made sure that the optimized structure fits into the rest of the model.
refine_model_mmtk.py -m my_model.pdb -c A -r 50-55 -y 1000\ -o optimized.pdb
what means that residues in range 50-55 from chain A of my_model.pdb will be optimized in 1000 cycles (if chain id is not specified, then the script assumes that chain A should be refined) and the entire structure containig optimized fragment will be saved to optimized.pdb. It has to be remembered, that residue numbers given after '-r' must correspond to residues in a linear sequence of RNA, and not to the numbering of residues in a PDB file (because a residue id can contain letter e.g. 20A).
It is also possible to optimize an entire model with refine_model_mmtk.py.
This may be necessary, e.g. to resolve conflicts between different parts of the chain.
Optimization takes considerably longer than for a single fragment (several hours for a tRNA-sized structure).
Example:
refine_model_mmtk.py -m my_model.pdb -y 1000\ -o optimized_fragment.pdb
Important notice! In this example a whole RNA chain is refined. But if the first residue contains an OP3 atom
(what is a characteristic feature of a first residue at the 5' end), then because of the bug in MMTK,
the program returns a Python IOError:
"IOError: Atom O3P of PDB residue RG not found in residue RG of object .RG_1". The solution is to exclude the first
residue at 5' end from the refinement, by running:
refine_model_mmtk.py -m my_model.pdb -y 1000 -r 2-78\ -o optimized_fragment.pdbwhere 78 corresponds to the 78th residue in a linear sequence - in this case the residue at RNA 3' end.
Modified nucleotides are not part of the standard AMBER forcefield. They are therefore excluded from the refinement by the MMTK script. A set of quantum mechanical parameters for 107 modified nucleotides has been published by (Aduri R et al. J Chem Theory Comput, 2007, 3(4), 1464–1475). They can be included in the AMBER parameter files to allow modifications to be refined in the same way.
|
| Copyright© - Adam Mickiewicz University - All rights reserved |