
| | |||||||||||||||||
| |||||||||||||||||
Tutorial
Introduction
This tutorial is a brief introduction to the capabilities of the ModeRNA tool - the program for comparative modeling of RNA. Here you'll see how to create homology models of two tRNA molecules. These will be models of:
Caution: Casual modelers must be warned that for large RNA molecules with complex structures, the development of a good alignment may require laborious manual preparation of the input data based on previous expertise of the respective RNA family.
Summary
In both cases the template for modeling will be E. coli threonyl tRNA (chain B from entry 1QF6). In the case of the first target, E. coli tRNA Tyr (QUC), there's a crystal structure of this tRNA in the PDB database - entry 1C0A, so you'll be able to compare the model to the native structure.
A model of Escherichia coli tRNA Tyr will be built automatically, but building of the tRNA from the second example will require more manual approach (because of clashes occuring in the model).
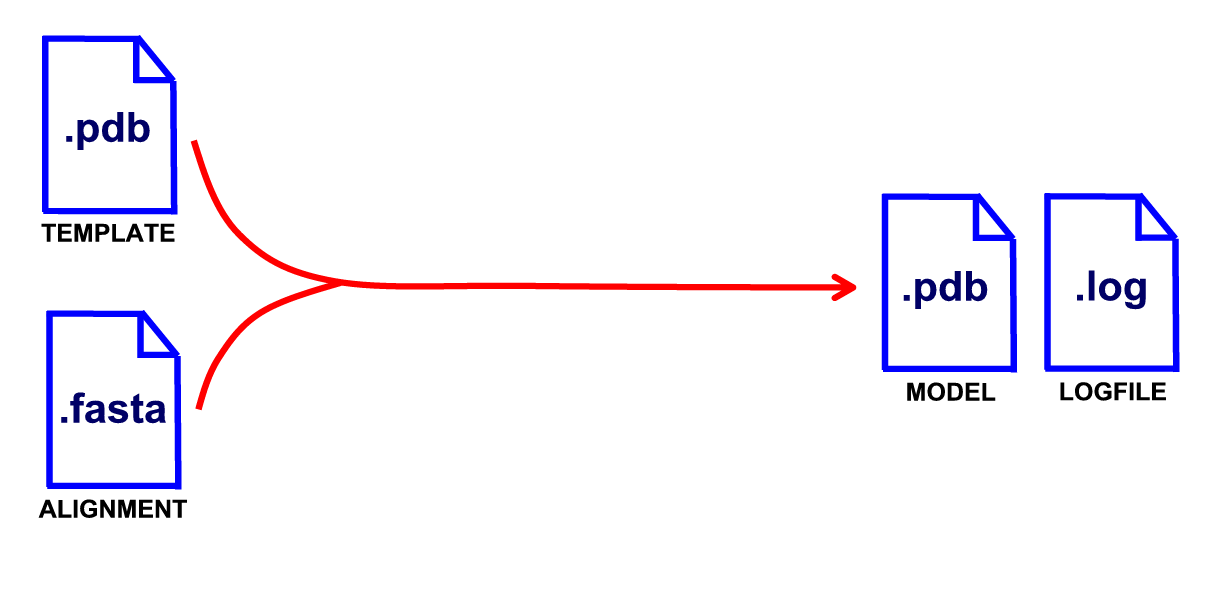
Checking the ModeRNA installation
Instructions on how to install ModeRNA can be found in the Installing section. Before we begin our adventure with comparative modeling of RNA, let's do some tests to make sure that ModeRNA works flawlessly on your machine.If you use the source distribution:
- Start the python interpreter and type:
>>> from moderna import *
If you use the Windows binary:
- Open a shell window.
Start -> Execute -> cmd - Go to the directory to which you unpacked ModeRNA. E.g.
cd Desktop\moderna
- Run the help function of the program:
moderna.exe -h
- If the above command does not return any errors, ModeRNA is ready for usage!
Now we're ready to move on to modeling tRNA Tyr (GUA) from Escherichia coli.
Modeling of E. coli tRNA Tyr (GUA)
Summary
We start by creating a directory for the project, let's name it tRNA_ASP. We'll keep all downloaded files and run ModeRNA there.
The template
Because we'll use the same template for modeling as we used in the case of tRNA Asp (chain B of 1QF6), you can use the preprocessed chain B of 1QF6 from the previous example or you can download if from here.
The alignment
In figure 2 you can see the alignment between the sequence of E. coli tRNA Tyr (its sequence was again taken from the MODOMICS database) and the sequence of a template (chain B from 1QF6).
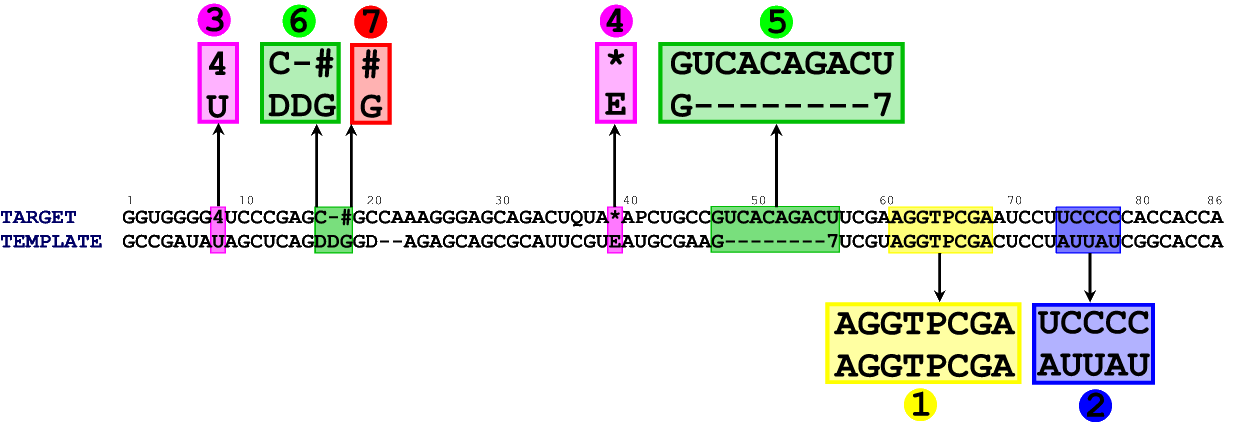
Figure 2. An alignment between target tRNA (chain B from 1QF6) and a template tRNA (E. coli tRNA Tyr). Although large part of the model can be built automatically (regions 1, 2, 3, 4, 6, 7), there is one part of the model which requires manual approach. A, C, U, G denote standard nucleotides, - denotes a gap. All other characters represent posttranscriptionally modified nucleotides. Explanation of the abbreviations can be found in the MODOMICS database.
Building the model automatically
When you've saved those files in tRNA_TYR directory, you can run ModeRNA. You can again use the fully automated mode, by e.g. typing in the Python interpreter:
from moderna import *
t = load_template('1QF6_B_tRNA.pdb', 'B')
a = load_alignment('aln_1QF6_E_coli_Tyr2_GUA.fasta')
m = create_model(t,a)
m.write_pdb_file('model_Tyr_E_coli.pdb')
These commands make ModeRNA create the moderna.log and model_Tyr_E_coli.pdb files in the tRNA_TYR directory.
Checking the correctness of the geometry
ModeRNA can check whether an RNA structure has any exceptional bond lengths, flat angles or dihedrals. This function is available from the Python interpreter only:
analyze_geometry(t)
analyze_geometry(m)
Modeling of E. coli tRNA Asp (QUC)
- The template structure
- The alignment
- Building the model automatically
- Model verification
- Model refinement
Summary
We will create a model for tRNA Asp from a very similar structure. The template for modeling will be E. coli threonyl tRNA (chain B from entry 1QF6).
As in the first example, we start by creating a directory for the project - let's name it tRNA_ASP - we'll store there all downloaded files.
The template structure
Then let's get a template for modeling - we'll use the tRNA from the record 1QF6 from the PDB database. Open 1QF6.pdb in any text editor - notice that chain B with tRNA contains also water molecules - these have to be removed. You can do it manually or, for example, in PyMol by selecting the RNA chain and saving it in a separate file (File -> Save Molecule...). If you want, you can download the preprocessed PDB file - 1QF6_B_tRNA.pdb.
For the modeling to start, we need to make sure that the template sequence is the same as in the alignment, so a good idea to start with is to get the sequence out of the structure. This can be done from the command line:
moderna.exe -t 1QF6_B_tRNA.pdb -c Bor
moderna.py -t 1QF6_B_tRNA.pdb -c B
The output should look like this:
TEMPLATE SEQUENCE: GCCGAUAUAGCUCAGDDGGDAGAGCAGCGCAUUCGUEAUGCGAAG7UCGUAGGTPCGACUCCUAUUAUCGGCACCA
The alignment
In figure 1 you can see an alignment between the sequence of our target (E. coli tRNA Asp) and the sequence of the template structure (chain B from 1QF6) . We took both sequences from the MODOMICS database.
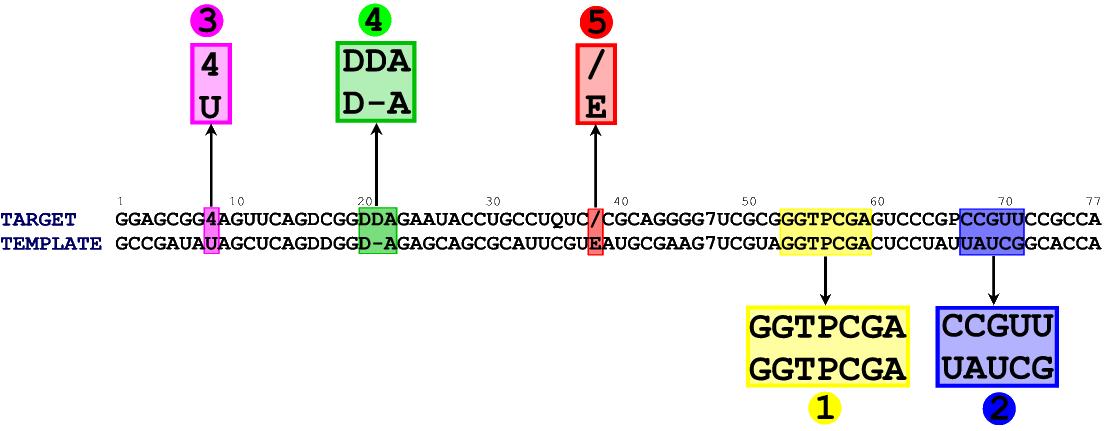
In the alignment file, both sequences are written in the FASTA format [download aln_1QF6_E_coli_1C0A.fasta]. Notice that in the alignment file the order of the sequences is important, as ModeRNA will treat the second one as a template, which will be used for modeling. You can check whether the template sequence is identical to the one generated above, but ModeRNA will do this for you automatically.
Building the model automatically
If you've saved the alignment file and the template in the tRNA_ASP folder, you are ready to build the model! When the template and a target sequence are similar (as in this case), an RNA model can be built automatically. This can be done in four ways:
a) From the command line (quick)
Run Moderna with the following arguments:
moderna.py -t 1QF6_B_tRNA.pdb -c B -a aln_1QF6_E_coli_1C0A.fasta\ -o model_Asp_E_coli.pdb
b) Using the Windows binary (also quick)
Run Moderna with the following arguments:
moderna.exe -t 1QF6_B_tRNA.pdb -c B -a aln_1QF6_E_coli_1C0A.fasta\ -o model_Asp_E_coli.pdb
c) Using the Python interpreter (detailed)
Type the following in the Python interpreter:
from moderna import *
t = load_template('1QF6_B_tRNA.pdb', 'B')
a = load_alignment('aln_1QF6_E_coli_1C0A.fasta')
m = create_model(t,a)
m.write_pdb_file('model_Asp_E_coli.pdb')
These commands should be self-explanatory. They tell the Python interpeter to:
- import ModerRNA's interface functions
- load template - that is chain B from 1QF6_B_tRNA.pdb
- load alignment file - that is alignment from aln_1QF6_E_coli_1C0A.fasta
- create the model
- save the model in tRNA_ASP directory in model_Asp_E_coli.pdb.
d) Running a Python script (also detailed)
Run the above commands as a simple script (e.g. example_Asp.py). Download it and on any UNIX platform type:
python example_Asp.py
Model verification
Inspecting the logfile
Besides creating model_Asp_E_coli.pdb, ModeRNA has also logged its activites to a file named moderna.log. They should be both in the tRNA_ASP directory (notice that if you run ModeRNA from Python interpreter, the data will be written to the logfile after closing the interpreter only). You can also download them from the Tutorial webpage without actually running ModeRNA on your computer (model_Asp_E_coli.pdb and moderna.log). If you check the logfile you'll see that the model has been successfuly built (near the end of the file, you can see: "Checking whether alignment matches with model. Sequences match:").
Checking for interatomic clashes
When the tRNA model is ready, a user can check it for steric clashes between its atoms. This procedure can be performed using built-in ModeRNA's functionality. If the user is working from the Python interpreter, clash recognition can be done by running the following commands:
cr = ClashRecognizer()
cr.find_clashes_in_pdb('model_Asp_E_coli.pdb')
In this example, there is one clash in model_Asp_E_coli.pdb - the find_clashes_in_pdb() method will report that there is a clash between residues 21 and 47 in this example. This problem can be solved by refining the model with MMTK program. List of all available methods of ClashRecognizer, can be obtained by typing:
help(cr)
where 'cr' is an instance of ClashRecognizer class.
Model verification
If you now open moderna.log, you'll see that in this case that ModeRNA was able to complete the modeling process.
After opening that file, you'll see the following message: "Checking whether alignment matches with model.
Sequences match:".
We're basically done. Of course, we can also validate the new RNA model, by typing:
analyze_geometry(t)
analyze_geometry(m)
Comparing the model to the real PDB structure
There is a model of the target E. coli tRNA Asp in the PDB database (1C0A). The only difference between the sequence of tRNA Asp from 1C0A record and its sequence from the MODOMICS database is that in the position 38 in the alignment in Figure 1 (first sequence, highlighted in red), the former contains a standard nucleotide A, while the latter contains / (2-methyladenosine). In general, data from the MODOMICS database is more accurate.
Model refinement (clash removal)
MMTK is a molecular dynamics toolkit written in Python. For that reason, it integrates well with ModeRNA. It implements a complete AMBER forcefield and parameters. If you have MMTK installed (if not check MMTK installation instructions), you can run a Conjugate Gradient minimization on the model obtained in the previous steps, to optimize local geometry - most of all, the ends of suboptimally inserted loops. To perform the refinement and remove steric clashes, type:
refine_model_mmtk.py -m model_Asp_E_coli.pdb -c A -y 250\ -r 2-77 -o optimized.pdb
However, MMTK has disadvantages, e.g. it is slower than other systems and more challenging to install. Possible alternatives include HyperChem (commercial), or a different implementation of the AMBER forcefield.
|
| Copyright© - Adam Mickiewicz University - All rights reserved |